Safety & Tolerability Profile
The safety of EMRELIS was evaluated in the LUMINOSITY study, which included 168 patients with locally advanced or metastatic EGFR wt NSq NSCLC with
c-Met protein overexpression who received EMRELIS.
Serious adverse reactions occurred in 35% of patients. Serious adverse reactions occurring in ≥2% of patients included ILD/pneumonitis (5%), pneumonia (5%), peripheral neuropathy (3.6%), and pleural effusion (2.4%). Fatal adverse reactions occurred in 5% of patients who received EMRELIS, including ILD/pneumonitis (1.8%), pneumonia (1.2%), sudden death (1.2%), noninfectious endocarditis (0.6%) and myocardial infarction (0.6%).1
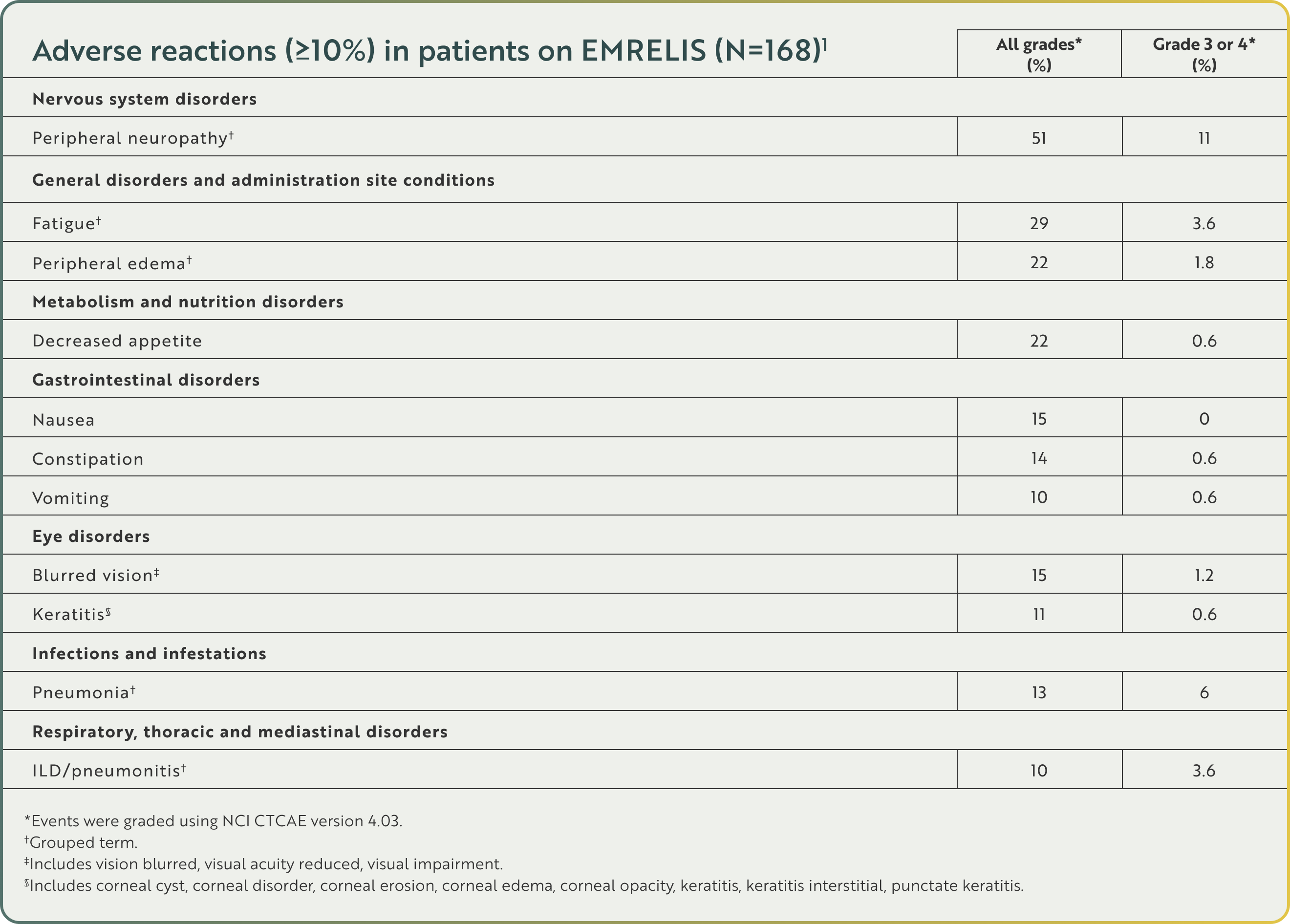
Adverse reactions (≥10%) in patients on EMRELIS (N=168)1
Nervous system disorders
All grades* (%) 51
Grade 3 or 4* (%) 11
General disorders and administration site conditions
All grades* (%) 29
Grade 3 or 4* (%) 3.6
All grades* (%) 22
Grade 3 or 4* (%) 1.8
Metabolism and nutrition disorders
All grades* (%) 22
Grade 3 or 4* (%) 0.6
Gastrointestinal disorders
All grades* (%) 15
Grade 3 or 4* (%) 0
All grades* (%) 14
Grade 3 or 4* (%) 0.6
All grades* (%) 10
Grade 3 or 4* (%) 0.6
Eye disorders
All grades* (%) 15
Grade 3 or 4* (%) 1.2
All grades* (%) 11
Grade 3 or 4* (%) 0.6
Infections and infestations
All grades* (%) 13
Grade 3 or 4* (%) 6
Respiratory, thoracic, and mediastinal disorders
All grades* (%) 10
Grade 3 or 4* (%) 3.6
*Events were graded using NCI CTCAE version 4.03.
†Grouped term.
‡Includes vision blurred, visual acuity reduced, visual impairment.
§Includes corneal cyst, corneal disorder, corneal erosion, corneal edema, corneal opacity, keratitis, keratitis interstitial, punctate keratitis.
Other clinically relevant adverse reactions in <10% of patients who received EMRELIS included arthralgia, dizziness, dry eye, infusion-related reaction, and photophobia.1
No baseline eye exam is required prior to initiating EMRELIS.
Discontinuations and dose reductions/interruptions1

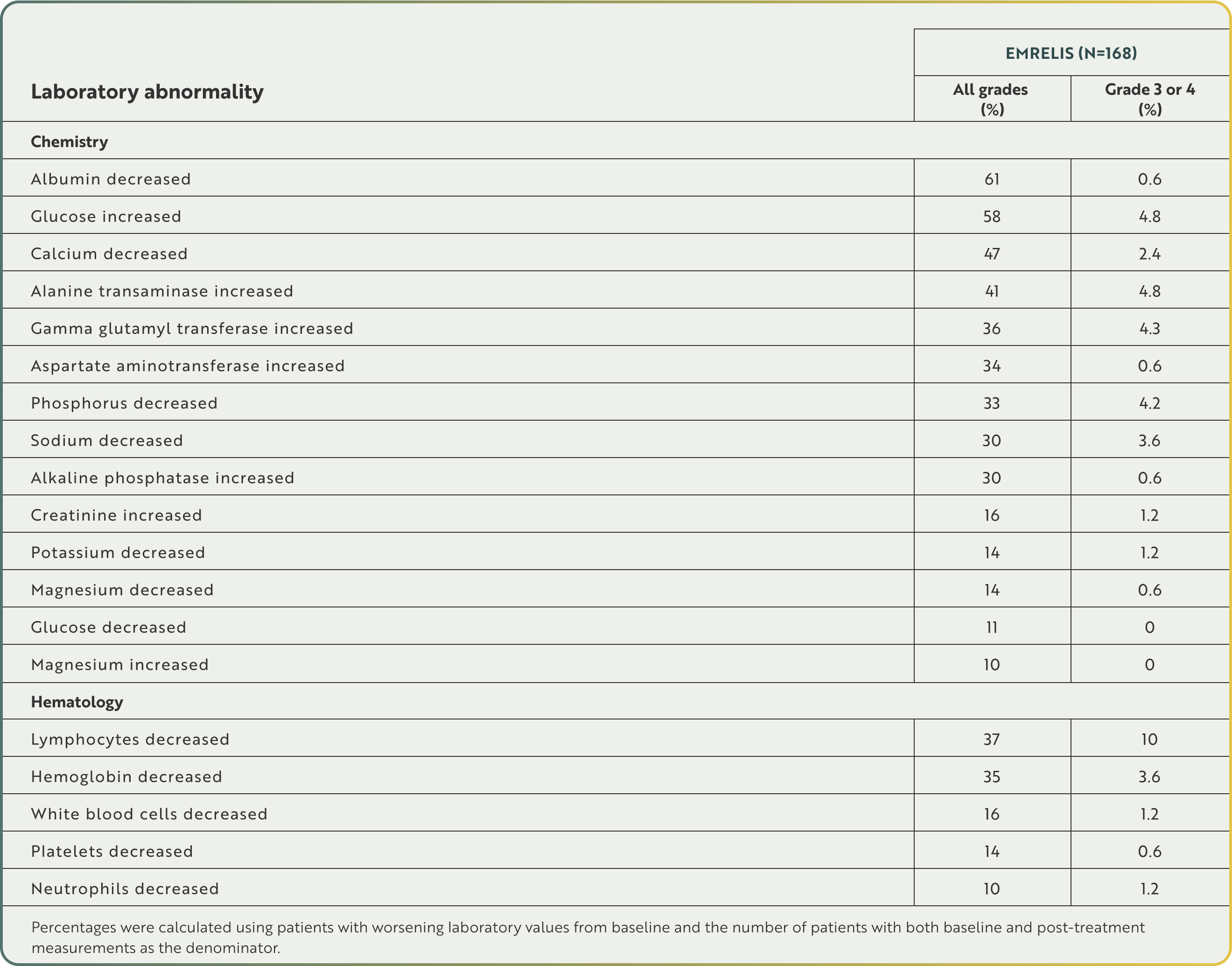
Permanent discontinuations of EMRELIS due to adverse reactions occurred in 30% of patients. Adverse reactions which resulted in permanent discontinuation of EMRELIS in ≥2% of patients included peripheral neuropathy and ILD/pneumonitis
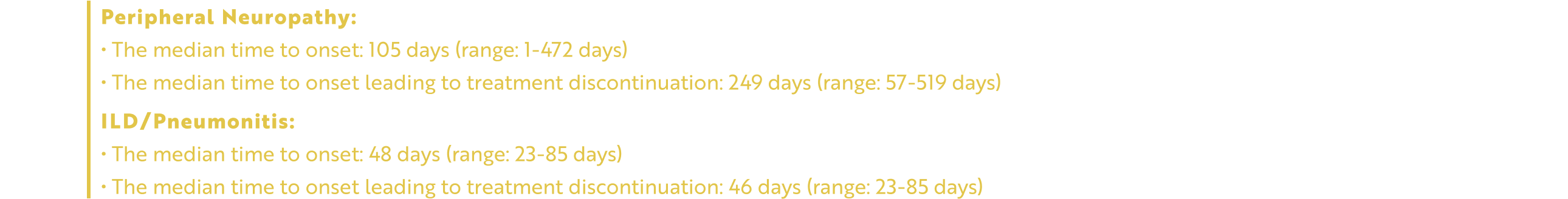

Dose reductions due to adverse reactions occurred in 28% of patients. Adverse reactions which required dose reductions in ≥2% of patients included peripheral neuropathy, fatigue, and keratitis

Dosage interruptions due to adverse reactions occurred in 44% of patients. Adverse reactions which required dosage interruption in ≥2% of patients included peripheral neuropathy, fatigue, pneumonia, increased ALT, blurred vision, COVID-19, ILD/pneumonitis, and keratitis
Warnings and Precautions1
Peripheral Neuropathy
EMRELIS can cause peripheral neuropathy, including peripheral sensory neuropathy and peripheral motor neuropathy. In the safety population, peripheral neuropathy occurred in 51% of patients treated with EMRELIS, including Grade 3 in 11%. These adverse reactions included peripheral sensory neuropathy in 45% of patients and peripheral motor neuropathy in 9%. The median time to onset of peripheral neuropathy was 105 days (range: 1 to 472 days). Peripheral neuropathy led to permanent discontinuation of EMRELIS in 13% of patients. The median time to onset of peripheral neuropathy leading to treatment discontinuation was 249 days (range: 57 to 519 days). Of the 7 patients with motor neuropathy ongoing as of their last dose of EMRELIS, 6 had persistent Grade 1 or 2 symptoms 30 days after their last dose.
Monitor patients for signs and symptoms of new or worsening peripheral neuropathy such as hypoesthesia, hyperesthesia, paresthesia, a burning sensation, neuropathic pain, or muscle weakness. Withhold, reduce the dose, or permanently discontinue EMRELIS based on severity.
Interstitial Lung Disease/Pneumonitis
EMRELIS can cause severe, life-threatening, or fatal interstitial lung disease (ILD)/pneumonitis. In the safety population, ILD/pneumonitis occurred in 10% of patients treated with EMRELIS, including Grade 3 in 3% and Grade 4 in 0.6%. There were 3 fatal cases of ILD/pneumonitis in patients who received EMRELIS. The median time to onset of ILD/pneumonitis was 48 days (range: 23 to 85 days). ILD/pneumonitis led to permanent discontinuation of EMRELIS in 7% of patients. The median time to onset of ILD/pneumonitis leading to treatment discontinuation was 46 days (range: 23 to 85 days).
Advise patients to immediately report cough, dyspnea, fever, and/or any new or worsening respiratory symptoms. Monitor patients for signs and symptoms of ILD/pneumonitis. Withhold or permanently discontinue EMRELIS based on severity.
Ocular Surface Disorders
EMRELIS can cause ocular surface disorders including blurred vision, visual impairment, keratitis, and dry eye. In the safety population, ocular surface disorders occurred in 25% of patients treated with EMRELIS. The most common ocular surface disorders were blurred vision (15%), keratitis (11%), and dry eye (5%). Grade 3 ocular surface disorders occurred in 1.2% of patients [blurred vision (1.2%), and keratitis (0.6%)]. The median time to onset of ocular surface disorders was 47 days (range: 1 to 319 days).
Monitor patients for ocular surface disorders during treatment with EMRELIS. Withhold EMRELIS and refer patients to an eye care professional for an ophthalmic examination and treatment for patients who develop Grade ≥2 ocular toxicity. Withhold or permanently discontinue EMRELIS based on severity.
Infusion-Related Reactions
EMRELIS can cause infusion-related reactions (IRR); signs and symptoms of IRR include dyspnea, flushing, chills, nausea, chest discomfort, and hypotension. The median time to onset of IRR was 28 days (range: 1 to 43 days). In the safety population, IRR occurred in 3% of patients treated with EMRELIS, including Grade 3 in 1.2% and Grade 4 in 0.6%. IRR led to permanent discontinuation of EMRELIS in 0.6% of patients.
Monitor patients for signs and symptoms of infusion reactions during EMRELIS infusion. Withhold, reduce the rate of infusion, or permanently discontinue EMRELIS based on severity. For patients who experience IRR, administer premedications prior to subsequent infusions.
Embryo-Fetal Toxicity
Based on the mechanism of action and findings in animals, EMRELIS can cause fetal harm when administered to a pregnant woman. The small molecule component of EMRELIS, monomethyl auristatin E (MMAE), administered to rats caused adverse developmental outcomes, including embryo-fetal mortality and structural abnormalities, at exposures similar to those occurring clinically at the recommended dose.
Advise patients of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with EMRELIS and for 2 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with EMRELIS and for 4 months after the last dose.
ALT=alanine transaminase; NCI CTCAE=National Cancer Institute Common Terminology Criteria for Adverse Events.
Next: